Campus News
Study reveals the molecular origin of the genetic disease cystinosis
New understanding of how the transporter protein cystinosin functions may lead to better treatments for a devastating genetic disease.

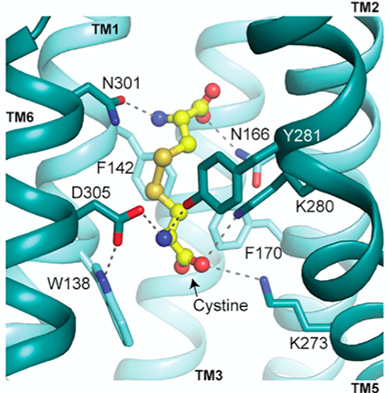
The rare genetic disease cystinosis is caused by mutations in the gene for a protein called cystinosin. A team of scientists has now solved the structure of cystinosin and determined how mutations interfere with its normal function, providing insights into the underlying mechanisms and suggesting a way to develop new treatments for the disease.
The new study, published September 15 in Cell, involved a collaborative effort by researchers at UC Santa Cruz, Stanford University, and the University of Texas Southwestern Medical Center, who combined their expertise in three specialized methods for studying protein structure and function: x-ray crystallography, cryogenic electron microscopy (cryo-EM), and double electron-electron resonance (DEER).
“This paper could set a model for how to combine those three areas, along with biochemical assays, to quickly narrow in on how a protein functions and identify a therapeutic strategy,” said Glenn Millhauser, distinguished professor and chair of chemistry and biochemistry at UC Santa Cruz and a corresponding author of the paper.
Cystinosin is a specialized transporter protein that plays a crucial role in how cells manage the essential amino acid cysteine. Cells are constantly recycling proteins, breaking them down into their constituent amino acids for use in building new proteins. Transporters like cystinosin move the amino acids out of lysosomes—the cellular compartments where proteins are broken down—into the cell to be reused. When cystinosin isn’t functioning properly due to mutations, a form of cysteine (a dimer called cystine) builds up inside the lysosomes.
The abnormal accumulation of cystine causes widespread damage to tissues and organs and can lead to kidney failure, muscle wasting, and other problems.
“It’s a rare disease, but it can be deadly,” Millhauser said. “If it’s untreated, people with cystinosis usually die by age ten.”
Cystinosin adopts different conformations when it is open to the inside of the lysosome to load cystine and when it is open to the outside to release cystine. The research teams at Stanford (led by Professor Liang Feng) and at UT Southwestern (led by Professor Xiaochun Li) solved the structures of cystinosin in these different structural conformations using x-ray crystallography and cryo-EM.
Understanding cystinosin’s structural changes through the transport process, however, required the DEER studies performed by Millhauser’s lab. DEER is a specialized magnetic resonance technique that can be used to determine how a protein changes its shape.
“With that we were able to figure out the mechanism that allows cystinosin to switch between those different states, and we could narrow in on which of the protein’s amino acids were driving the transition,” Millhauser said. “Now we can see how the mutations are changing the protein’s ability to change shape and pump cystine out of the lysosome.”
These new insights into the molecular mechanics of cystinosin’s transport activity not only provide a more detailed understanding of the pathogenesis of cystinosis, but also suggest a possible therapeutic strategy to treat the disease. “It may be possible to enhance the transport activity of cystinosin by developing conformation-selective small molecules or biologics that favor a cytosol-open conformation,” the authors wrote.
A similar approach could be used to target other transporter proteins, which are involved in a wide range of diseases.
The authors of the paper include co-first authors Tufa Assafa at UC Santa Cruz, Xue Guo at Stanford, and Philip Schmiege at UT Southwestern, and coauthors Yan Xu at Stanford, Rong Wang, Linda Donnelly, and Michael Fine at UT Southwestern, and Xiaodan Ni and Jiansen Jiang at the National Heart, Lung, and Blood Institute. This work was funded in part by the National Institutes of Health.