Campus News
The team behind a tree of 10 million Covid sequences
10 million sequences of COVID-19’s genomic code have now been organized into a phylogenetic tree in the UC Santa Cruz SARS-CoV-2 Browser, which is the largest tree of genomic sequences of a single species ever assembled.

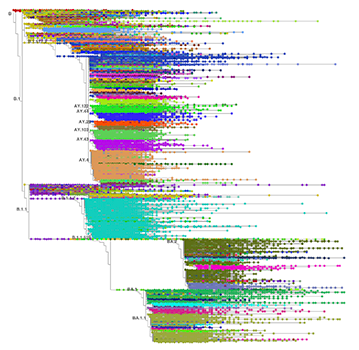
10 million sequences of COVID-19’s genomic code have now been organized into a phylogenetic tree in the UC Santa Cruz SARS-CoV-2 Browser, which is the largest tree of genomic sequences of a single species ever assembled. This accomplishment is impressive for both the computer engineering feat of processing such a massive amount of data and the incredible dedication and coordination of the researchers involved.
“It is an astounding thing that has happened there,” said Clay Fischer, Project Manager for the UCSC Genome Browser.
All of these sequences are assembled by the researchers into a phylogenetic tree that shows the evolutionary history of the virus, with different branches representing the lineages that have mutated throughout the pandemic. This tree is powered by a software tool called UShER that was developed at the UC Santa Cruz Genomics Institute and is hosted on the UCSC Genome Browser website.
Many hands from around the world have brought the Genomics Institute these 10 million sequences that live on the UShER tree. Clinicians worldwide have administered tests to be sent off to local labs, which then sent the samples on for sequencing. Once they are sequenced, they become digital files that are uploaded to databases for genomic information such as GISAID, GenBank, or the COG-UK database.
Angie Hinrichs, a senior software architect at the UCSC Genome Browser and self-described “data wrangler,” built a pipeline to pull these sequences into the UShER tree automatically. But this process was complicated as some databases, like GISAID, had restrictions that necessitated the manual download of sequences.
“For the first half of 2021, I would download them every night before bed,” Hinrichs said.
Hinrichs has worked at the UCSC Genome Browser for twenty years. She keeps a low profile, usually preferring to work behind the scenes than in the spotlight. But according to her colleagues, her work curating the tree of COVID-19 genomes and coordinating with the CDC and other health organizations has been of great importance to the pandemic relief effort. She is a part of the Pango team of volunteers who have been monitoring virus sequences to identify new variants. She takes on the ongoing, daily maintenance of updating and annotating the UShER tree, which recently became the default software used by the Pangolin tool, a system used by health officials worldwide to track the spread of variants in their community.
UShER was created early in the pandemic, when researchers at the UC Santa Cruz Genomics Institute recognized that tracing the evolution of a quickly evolving global pathogen like COVID-19 would require a phylogenetic tree that was able to handle an unprecedented amount of data. So, the Genomics Institute’s scientific director David Haussler gathered together a team to focus on pathogen genomics, led by Assistant Professor of Biomolecular Engineering Russell Corbett-Detig and including then-postdoc Yatish Turakhia. Turakhia originally wrote the UShER software, which has the ability to rapidly add a new genome sequence to a very large tree of genome sequences.
Making a tree that can handle so much data is an incredible feat of computer engineering that has required herculean efforts from a number of researchers. Before the current pandemic, phylogenetic trees for comparing viral samples were relatively common, but they were built from comparatively small numbers of sequences.
As unprecedented numbers of SARS-CoV-2 sequences became available, the standard tree-building tools simply could not keep up, and researchers often struggled to make sure their analysis kept pace with the amount of samples they would receive. UShER’s software and the sustained effort of the team made it possible to grow the tree apace with the pandemic’s flood of sequences.
Hinrichs says that her two decades of experience working with the massive amounts of data stored on the UCSC Genome Browser helped prepare her to work with the COVID-19 lineages on UShER.
“This data coordination is what makes our resources really powerful,” Hinrichs said. “We have really great resources here, and really great people.”
One of those great resources is UCSC’s amazing computing hardware maintained by Jorge Garcia, Haifang Telc, and Erich Weiler. Hinrichs explained that having that computing power has been essential for this project.
“Big data is our thing, so we were ready to jump on this,” she said.
At the beginning of the pandemic, the UCSC pathogen genomics team made guesses as to how many COVID-19 sequences the tree would need to be able to handle. Only Corbett-Detig thought it would reach a million – no one anticipated reaching 10 million.
“I still get surprised at how far we’ve come,” Turakhia said. “The unimaginable amount of data we were able to handle and the fact that we are able to make sense of it quickly is mind-boggling as a computational genomicist.”
As the tree has grown, it has required constant attention and updates. Cheng Ye, an undergrad in Turakhia’s new lab at UC San Diego, was also able to figure out a way to add new sequences faster when the tree had grown to contain millions of sequences already, and helped develop a tool called MatOptimize that moves sequences around on the tree when more data makes it apparent that the original placement was less optimal.
Accumulating reliable data has been instrumental to better understanding what we are up against in the fight against COVID-19 and all its variants. While little was known about this virus at the start of the pandemic, the tree-building tools developed at UC Santa Cruz have helped to put the history of the virus in some perspective and to predict its future, and researchers across campus have leveraged their expertise to aid in the relief efforts. The progress has been astounding; but for the researchers on the browser team, the urgency of their mission and the sheer amount of data that needs to be curated has also been overwhelming at times. Fischer acknowledges that this level of dedication comes at a cost.
“It has been two years of blood, sweat, and tears,” he said.