Campus News
Prion study reveals how abnormal proteins damage nerve cells in the brain
Research on the prion protein may explain why nerve cells degenerate in prion diseases such as Creutzfeldt-Jakob disease.
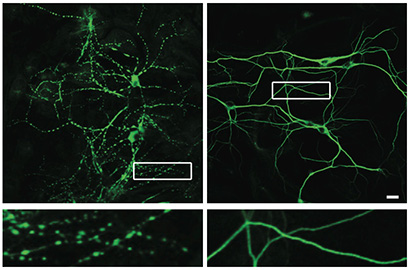
A new study has uncovered a molecular mechanism in the prion protein that may explain why nerve cells degenerate in prion diseases such as Creutzfeldt-Jakob disease (CJD).
The findings, which appear in the journal eLife, may one day lead to better therapies and treatments for these diseases.
Prion diseases, including CJD in humans and “mad cow disease” in cattle, are fatal neurodegenerative brain disorders caused by a misfolded form of the normal cellular prion protein. Other, more common neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, and frontotemporal dementia have also been found to involve abnormal accumulation of misfolded protein aggregates in the brain.
According to the researchers, how nerve cells are damaged in prion diseases has remained a mystery until now. The new research combined cellular studies, led by Dr. David Harris at Boston University School of Medicine (BUSM), with structural analysis of prion proteins performed at UC Santa Cruz using nuclear magnetic resonance (NMR).
“Our work shows that the prion protein acts like a molecular on-off switch. In the ‘on’ positon, one end of the protein delivers a toxic signal to nerve cells, while in the ‘off’ position, the other end of the protein serves as a brake to reduce the toxic signal,” said Harris, corresponding author of the paper and professor and chair of biochemistry at BUSM. “This novel mechanism, in which the two parts of the prion protein have opposing functions, had not been fully appreciated before.”
The study also showed that copper, a normal constituent of brain chemistry, promotes interaction between the two ends, biasing the prion protein toward the “off” state. Disruption of this interaction led to degenerative changes in nerve cells.
“These data provide the best picture yet of the prion protein’s ‘off’ state,” said coauthor Glenn Millhauser, professor of chemistry and biochemistry at UC Santa Cruz. “Recognizing the nature of this state provides a platform for developing drugs to treat neurodegenerative diseases. This is an exciting time in prion biology.”
Using a multi-disciplinary approach involving electrophysiological, cellular, and biophysical techniques, the researchers found that parts of the prion protein lacking the “brake” region produced abnormal electrical currents in cells. Antibodies that interfered with the functioning of the brake region did the same. Importantly, the antibody treatment also caused severe degeneration of nerve cell dendrites, the regions that are essential for normal communication between nerve cells. In collaboration with Millhauser and others at UC Santa Cruz, the researchers applied a sophisticated chemical technique to demonstrate that the two ends of the prion protein interact with each to alter the amount of toxic signal that is delivered.
As a result of their findings, the researchers caution against administering antibodies against the prion as a possible therapy for both prion and Alzheimer’s diseases. “Our study sounds a serious warning about the possible detrimental side effects of this strategy, since we have shown that such antibodies cause dramatic degeneration of nerve cells by interfering with the normal on-off function of the prion protein,” Harris said.
The researchers hope their study will lead to better therapies for neurodegenerative disorders, as well as help clinicians avoid the possible dangerous side-effects of using anti-prion protein antibodies for therapeutic purposes.
Funding for this study was provided by the National Institutes of Health, the National Science Foundation, and the German Research Foundation.