Campus News
New responsible data sharing technique will enable better understanding of disease-causing genetic variants
Scientists may better understand and test for the genetic variations that cause cancer and other heritable diseases through the application of a novel strategy for securely sharing and analyzing genomic data developed at the UC Santa Cruz Genomics Institute.

Scientists may better understand and test for the genetic variations that cause cancer and other heritable diseases through the application of a novel strategy for securely sharing and analyzing genomic data developed at the UC Santa Cruz Genomics Institute.
Understanding the clinical significance of rare genetic variants requires analyzing large amounts of genomic and clinical data. Privacy policies, however, restrict the sharing of this information between institutions, and no single institution is likely to have all the resources needed for a robust analysis.
In a paper published March 9 in the journal Cell Genomics, UCSC researchers showed that an approach called federated analysis can overcome this problem by “bringing the code to the data.” This is the first application of federated analysis to enable classification of previously unclassified genetic variants.
“We have to find ways to get to the data that respect privacy, but still let researchers do their research, so the federated model is definitely the way of the future,” said James Casaletto, a PhD candidate at UCSC’s Baskin School of Engineering and the paper’s lead author.
The study focused on genetic variants of the breast cancer genes BRCA1 and BRCA2. People who inherit harmful variants of one of these genes have increased risks of breast, ovarian and other cancers. Many people, however, have variants of unknown significance (VUS) in these genes, meaning scientists don’t know if these variants are harmful or not.

The new study provides a more nuanced understanding of BRCA1 and BRCA2 variants. It also serves as a “proof of concept” of a novel data sharing and analysis technique for assessing the clinical implications of genetic variants.
Specific VUS are individually rare but collectively it is common for VUS in general to occur in the human population. To better understand exactly which VUS are disease-causing, researchers need to perform delicate analysis on a wide set of data, which can then be interpreted by experts to make clinical conclusions.
“This has to do with the everyday person who maybe wonders if there’s a history of cancer in her family, and if she’s inherited that family cancer risk,” said Melissa Cline, a research scientist at the UCSC Genomics Institute and co-author on the paper. “All of this work is going to the aim of making genetic testing better.”
However, most of the world’s human health data is ‘siloed’, or stored inaccessibly due to privacy laws, and institutions may be prohibited from exporting genomic data they collect, making it inaccessible to researchers who study genetic variants.
Additionally, engineering the software needed to execute these analyses is complex and usually cannot be undertaken by the average geneticist. UCSC researchers are addressing these two problems with their novel approach of federated analysis.
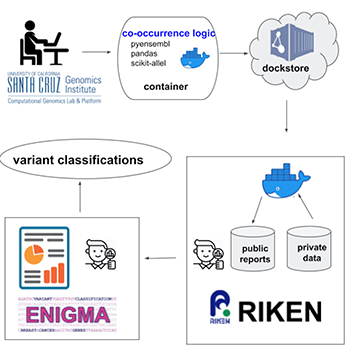
In the federated analysis approach, researchers bring the code to the data, avoiding the need to export sensitive data at all. UCSC Genomics Institute software is sent in a “container” to any collaborating institution around the world that is home to a valuable but protected set of genomic data. The collaborating institution then uses the software to analyze their data within their institution’s secure environment, generating summary data that does not reveal personal information about individual patients.
This approach ensures that patient-level data meets the strict privacy rules of an institution that do not allow them to export data, but allows researchers to collect a much wider pool of genomic data which can lead to better clinical conclusions. Moreover, federated analysis can get around the issues of uploading, downloading and moving around huge data sets that can be prohibitively large.
“[The paper is] a proof of concept that we have this container technology, we’ve leveraged it for BRCA1 and BRCA2, we’ve also demonstrated in the research that it can be used for other genes – genotypes and phenotypes,” Casaletto said.
For this project, UCSC researchers collaborated with the RIKEN Center for Integrative Medical Sciences in Japan to analyze their biobank of BRCA1 and BRCA2 genomic data. These genes are inherited from a person’s parents and when mutated can lead to an increased risk of breast, ovarian, and other cancers.
They used this to make discoveries about which specific variants in the BRCA1 and BRCA2 genes led to cancer and which left patients unaffected, moving the needle on a number of previously uncertain variants.
In undertaking this analysis, the researchers were able to help address the lack of diversity in genetic databases.
“The genetics of white people are highly over-represented, the genetics of non-white people are much more of a mystery, due to a lot of historical biases in data collection,” Cline said. “We were also able to add together a little more knowledge on Japanese genetics than was previously available.”
Further collaboration using federated analysis with institutes worldwide could similarly do much to address the lack of representation of non-white people and empower institutions that may be resource-poor to contribute to the global genomic data pool.
“What’s been done in the past is basically a lot less data sharing, so the name of the game is really global data sharing,” Cline said.
The researchers also work with the The Global Alliance for Genomics and Health (GA4GH), which helps set policy and create technical standards for responsible, ethical data sharing. They gave guidance on what data sharing can be done legally and responsibly, and helped establish methods to make the Genomics Institute software portable across different operating systems and environments to allow for collaboration like that in this project.
Other coauthors of the paper include Charles Markello of UCSC, Michael Parsons and Amanda Spurdle of Australia’s Berghofer Medical Research Institute, and Yusuke Iwasaki and Yukihide Momozawa of Japan’s RIKEN Center for Integrative Medical Sciences.