Campus News
UC Santa Cruz to lead effort to build a new map of human genetic variation
The Simons Foundation awarded UCSC up to $1 million to develop a comprehensive Human Genome Variation Map for scientific and medical research

Researchers at the UC Santa Cruz Genomics Institute have received a grant for up to $1 million from the Simons Foundation to develop a comprehensive map of human genetic variation. The Human Genome Variation Map will be a critical new resource for both medical research and basic research in the life sciences.
The one-year pilot project aims to overcome the limitations of the current model for analyzing human genome data, which is based on the use of a single reference sequence for the human genome. Essentially, all novel sequencing data is analyzed by mapping new genome sequences to this one reference set of 24 human chromosomes to identify variants. But this approach leads to biases and mapping ambiguities, and some variants simply cannot be described with respect to the reference genome, according to David Haussler, professor of biomolecular engineering and director of the Genomics Institute at UC Santa Cruz.
“One exemplary human genome cannot represent humanity as a whole, and the scientific community has not been able to agree on a single precise method to refer to and represent human genome variants. There is a great deal we still don’t know about human genetic variation because of these problems,” said Haussler, who will lead the project with co-investigator Benedict Paten, a research scientist at the Genomics Institute.
“Tower of Babel”
According to Paten, the proliferation of different genomic databases has resulted in hundreds of specialized coordinate systems and nomenclatures for describing human genetic variation. UC Santa Cruz genomics researchers are intimately familiar with this “Tower of Babel” of databases through their work to display data from all these sources on the widely used UCSC Genome Browser. Launched in July 2000 shortly after UC Santa Cruz posted the first working draft of the human genome sequence on the Internet, the browser now serves 130,000 researchers around the world and gets more than 1 million web page requests per day.
“For now, all our browser staff can do is to serve the data from these disparate sources in their native, mutually incompatible formats,” Paten said. “This lack of comprehensive integration, coupled with the over-simplicity of the reference model, seriously impedes progress in the science of genomics and its use in medicine.”
Recently, with funding from the Simons Foundation, researchers David Reich and Nick Patterson at the Broad Institute of MIT and Harvard have amassed more than 300 complete human genome sequences representing a range of ethnicities. Haussler and Paten plan to use this set of human genomes, which they say is deeper and more completely organized than any prior human data set, to build a new graph-based human reference genome structure.
“This unique data set of genome diversity gives us an opportunity to define a comprehensive reference genome structure that can be truly representative of human variation. Eventually, we will want to expand it to include many more genomes, but this pilot project will focus on building a map structure based on the Reich-Patterson data set,” Paten said.
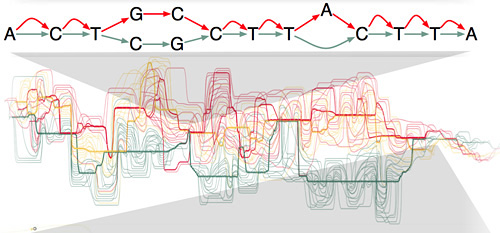
The new Human Genome Variation Map will replace the current snarl of isolated, incompatible databases of human genetic variation with a single, fundamental representation formalized as a very large mathematical graph. The clean mathematical formulation is a major strength of this new approach, Paten said.
The primary reference genome is a linear sequence of DNA bases (represented by the letters A, C, T, and G). To build the Human Genome Variation Map, each new genome will be merged into the reference genome at the points where it matches the primary sequence, with variations appearing as additional alternate paths along the genome. The resulting map will include all forms of human genome variation.
Global Alliance
The project dovetails with Haussler’s efforts as a leader of the Global Alliance for Genomics & Health (GA4GH), which involves more than 200 collaborating institutions that have agreed to work together to enable secure sharing of genomic and clinical data. The overall vision of the global alliance includes a genomics platform consisting of something akin to the planned Human Genome Variation Map, along with open-source software tools to enable researchers to mine the data for new scientific and medical breakthroughs.
The pilot project funded by the Simons Foundation grant is an essential first step in achieving this goal. The UC Santa Cruz team will collaborate with leading genomics researchers at other institutions to develop algorithms and formulate the best mathematical approach for constructing the Human Genome Variation Map. Initial work on developing a standard data model for the map is already under way in the context of the GA4GH Reference Variation Task Team co-chaired by Paten.
“We are bringing together the best people in the world to create and test different approaches for constructing the map. The first six months will be spent testing different algorithms on the trickiest regions of the genome,” Paten said.
By the end of the year, he expects to have a draft Human Genome Variation Map based on the full set of genomes. Paten and Haussler have also outlined the follow-up activities needed to extend the pilot project and fully realize their vision for the new map. Collaborators include scientists at major biomedical research institutions such as the Broad Institute, Memorial Sloan Kettering Cancer Center, UC San Francisco, Oxford University, the Wellcome Trust Sanger Institute in the U.K., and the European Bioinformatics Institute.
For medical researchers, the new map will make it easier to detect and analyze both simple and genomically complex variants that contribute to conditions that have a hereditary component, such as autism and diabetes. The map will also be a valuable tool for understanding recent human evolution, including the evolution and contribution to human diversity of hard-to-map DNA sequences such as mobile DNA elements.