Campus News
Ulcer-causing bacteria tamed by defect in cell-targeting ability
Without the ability to swim to their targets in the stomach, ulcer-causing bacteria do not cause the inflammation of the stomach lining that leads to ulcers and stomach cancer.
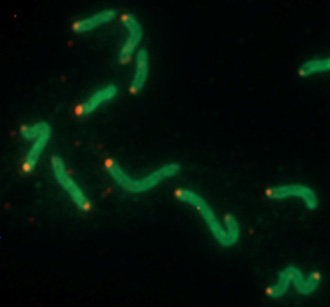
Without the ability to swim to their targets in the stomach, ulcer-causing bacteria do not cause the inflammation of the stomach lining that leads to ulcers and stomach cancer, according to a new study by researchers at the University of California, Santa Cruz.
The findings, published in the Proceedings of the National Academy of Sciences (Online Early Edition, week of Nov. 21-25), provide new clues about how the bacteria, called Helicobacter pylori, trigger harmful inflammation in some people. About half of all people worldwide are infected with H. pylori, but in most cases the infection does not cause any problems. Severe inflammation leading to ulcers or cancer occurs in only about ten percent of infections.
“If we can understand the pathways that cause the infection to go to this bad state of chronic inflammation, we may eventually be able to design treatments that would limit it,” said Karen Ottemann, a professor of microbiology and environmental toxicology at UC Santa Cruz and senior author of the PNAS paper.
Ottemann has been studying H. pylori chemotaxis, which is the bacteria’s ability to respond to specific chemicals in its environment by swimming toward or away from them. Her lab has developed a strain of the bacteria that is missing a single gene essential for chemotaxis. These defective bacteria cause much less inflammation than normal strains, even though they seem to have little trouble establishing infections in the stomach.
In the new study, the researchers looked at how the immune system responds to infections with normal and mutant strains of the bacteria. Their findings highlight the role of a particular type of white blood cell known as T-helper cell type 17 (Th17). Th17 cells promote chronic inflammation, but the researchers found that these cells were missing in the immune response to infection with the mutant strain.
The connection between chemotaxis and the immune response involves several steps. Previous work by other researchers has shown that Th17 cells respond to the combination of bacterial infection and dying host cells. Ottemann’s group found that the mutant strain of H. pylori causes much less cell death than normal strains. The researchers hypothesize that without chemotaxis, the mutant strains are not able to get close enough to the cells lining the stomach to deliver the bacterial toxins that induce cell death. The toxins trigger a process called apoptosis, a suicide program built into all cells and triggered by certain types of cell damage.
“The bacteria use chemotaxis to get close to the host stomach cells, and then they deliver packages of nasty molecules that kill host cells,” Ottemann said. “Previously, people thought the bacteria have to bind to the stomach cells. But it turns out they just have to be close enough to hit the cells with the cell-killing molecules. We think one reason they have the ability to swim is to hover close to their target cells.”
The missing gene in the mutant strain, called CheY, provides a link between the bacteria’s chemical sensors and their swimming mechanism, a whip-like flagellum that propels the spiral-shaped bacteria. The mutant bacteria can still swim, but they move aimlessly. “They’ve lost the connection between the sensory input and the behavior, so they just swim blindly,” Ottemann said.
H. pylori infections can be cured by taking antibiotics, but some studies have indicated that the infection may actually have some beneficial effects, at least for people who don’t get ulcers or stomach cancer. For example, H. pylori infection seems to reduce the chances of getting esophageal cancer. Some doctors have argued that controlling the negative effects of the infection may be preferable to eliminating it with antibiotics.
“The idea is that our bodies have adapted to it, and in 90 percent of people the bacteria act like a normal part of the body’s flora,” Ottemann said. “So the best thing might be to keep H. pylori in the stomach, but tame it so it wouldn’t cause inflammation. It’s possible we could tame it by targeting chemotaxis.”
Annah Rolig, a graduate student in molecular, cell, and developmental biology at UC Santa Cruz, is the first author of the PNAS paper. Coauthor J. Elliot Carter is at the University of South Alabama College of Medicine. This work was supported by a grant from the National Institutes of Health.